The case for automation
As demand for genomic testing increases, driven by greater accessibility and falling sequencing costs, genomic analysts are encountering increasingly complex patients in addition to their routine cases. While 70% of causal variants recur in multiple patients, the time needed to review previously un-encountered genes and variants has meant that a routine clinical analysis can now take up to 11 hours.
To continue providing high quality services for rare disease cases - and wider afield as the demand for genomic testing becomes more ‘mainstream’ - standardized tools to support variant interpretation are needed to continue successfully delivering genomics medicine services across the globe.
Congenica can reduce time spent on complex genetic analysis by over 90%
The release of Congenica 3.1 forms part of our ongoing commitment to embedding variant analysis best practices in an automated platform and intuitive workflow.

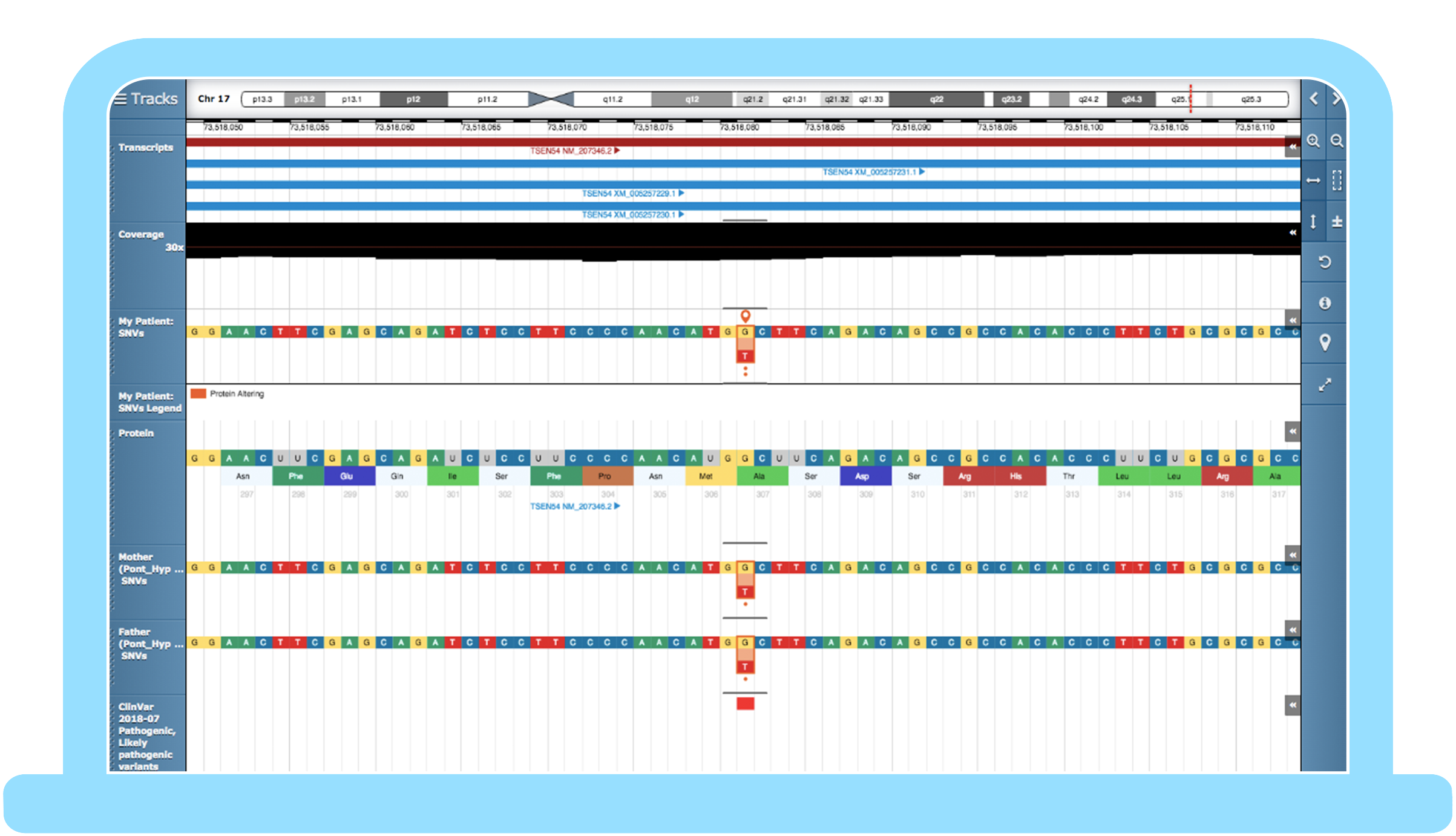
Congenica 3.1 – automating best practice in your workflow
Step 1: Set up a hereditary genomic test
The ability to appropriately filter variant data based on phenotype, family history, and variant attributes to quickly identify likely causal variants for additional clinical review is crucial.
When using whole exome sequencing or whole genome sequencing, Congenica allows you to set a programmable virtual gene panel for your test that includes all genes from your assay or just target genes, then implement your interpretation procedure using customizable, savable filters. If the initial panel test is inconclusive, our software enables you to perform subsequent panel-agnostic follow-on analysis, the full assay such as an exome or genome.
Automation supported by best-in-class analytics tool suite, built for solving the toughest challenges of whole genome sequencing at the scale of 10,000s of genomes
Step 2: “Have I seen this variant before?”
- If the answer is yes and you have the knowledgebase curated variant list set up for your project, Congenica Express will automatically apply your previous evidence to classify the known variant.
- If it’s a new variant not seen before, Congenica’s augmented analysis workflow expedites interpretation.
Step 3: Starting rare disease analysis
The initial analysis steps are completed by Congenica Exomiser, which automates variant prioritization in all sample types from genomes to panels. It takes into account any given variant in a sample to analyze the following:
- Rare (population allele frequency)
- Predicted consequences on protein (coding)
- Previously classified with confidence (e.g. ClinVar 2)
- Predicted Pathogenicity by in-silico predictors
- Match MOI hypothesis in family (e.g. unaffected parents = de novo)
- Does gene match my phenotype?
- How likely is this variant problematic combining all of the above evidence (Exomiser Combined Score) and additional predictive annotations.
This means that the most relevant variants and genes are automatically prioritized for analysis. In a set of random positively selected samples from the UK’s 100k Genome Project, Exomiser in Congenica accurately ranked causal variants in the top 5 for 92% of cases* – saving users time by reducing the burden of interpreting less-relevant variants.
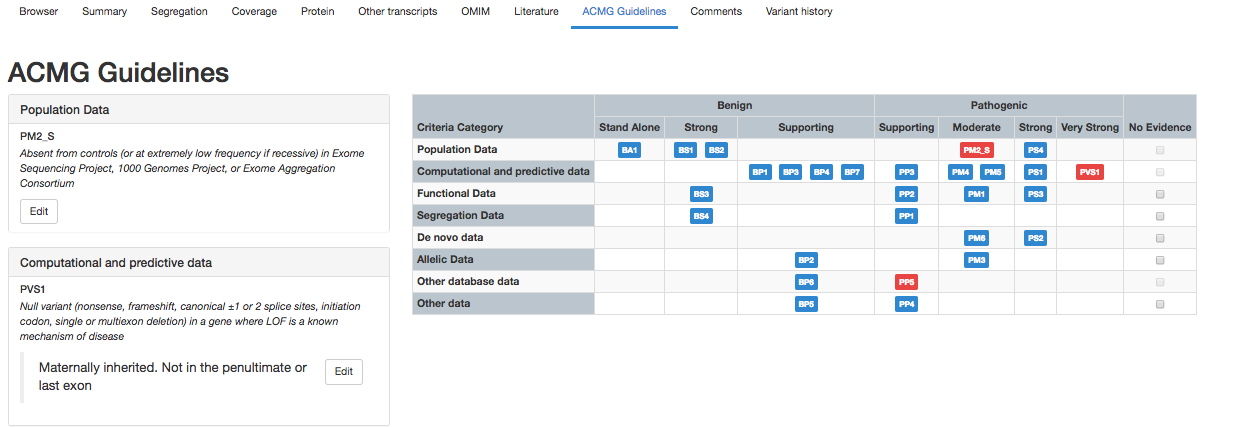
Step 4: Automated ACMG classification
Following variant and gene filtering, interpretation guidelines require the application of ACMG criteria and evidence to determine pathogenic variants. Congenica automatically provides over half of the ACMG criteria and evidence for the top Exomiser-ranked variants.
The results are delivered in sortable columns, detailing which variants are pathogenic, and criteria associated with the evidence. This combined workflow automatically presents the relevant variants and evidence so you can rapidly see whether your case has any likely pathogenic variants in genes associated with the phenotype. In internal assessments, the ACMG automation can save analysts ~30% of time (or about 4 minutes) per variant – with the typical exome case requiring the interpretation of 5-8 variants*.
Congenica 3.1 saves ~4 minutes per variant, potentially saving hours per case depending on complexity and lab SOPs
Best practice guidance stipulates collation and curation of data from numerous resources to support variant interpretation. The Congenica platform provides users with the resources recommended by the American College of Medical Genetics (ACMG) and ClinGen for genomic interpretation as well as supplementary annotations and resources for rapid filtering and analysis of both sequence (e.g. SNVs, Indels) and structural (e.g. CNVs) variants.
Congenica monitors global best practices, regulation, and scientific discovery to deliver a steady stream of product improvements to enable genomic medicine now and for the future.
Genomic data interpretation
While automation delivers significant time and quality benefits in the filtering and classification of variants, clinical expertise remains an essential part of the process. Your expertise is needed to contextualise the ACMG criteria and variant classification (cited as pain point in AMP 2021 genomic interpretation survey). The clinical knowledgebase within Congenica is loaded by users via Curated Variant List and takes precedence in automated analysis.
Setting standards in automated variant analysis
Importantly, manual retrieval of evidence according to these guidelines would be time-consuming, untraceable and prone to error. Collation of the increasing number of variant and disease datasets and annotation resources required, according to best practice, makes a clinical decision support system an ever-more essential tool to aid clinical variant interpretation.
To find out how the latest version of our software can help you, contact us today for a demo.
*https://cnfl.extge.co.uk/pages/viewpage.action?pageId=128355594


