
Life-Changing Answers - Congenica Genomics Clinical Case Series
This case study reveals how clinicians used Congenica genomic data analysis software to support their decision making and diagnose an exceptionally rare condition, CALFAN Syndrome. The results provided a single unifying diagnosis for a child's seemingly separate medical issues after years of investigation and uncertainty, avoiding unnecessary further procedures and testing.

CALFAN syndrome is an extremely rare disease that was first reported in 2015. Since then there have been fewer than 20 diagnoses of the disease. The disease phenotype is highly variable and is characterized by low γ-glutamyl-transferase cholestasis, acute liver failure, and neuro-degeneration. It can also include development or language delay and later onset of variable neurologic features.
The condition is caused by biallelic mutations in the SCYL1 gene, a gene that encodes for N-terminal kinase-like protein that is involved in intracellular transport processes.
Clinical Background
A 5 ½ year-old boy, the third child of first cousin parents, had normal development until he was 11 months old. A viral illness then led to developmental regression with unsteady walking. In addition there was poor growth, microcephaly, dysplastic corpus callosum, episodic liver decompensation (now resolving). Previous investigations including paediatric neurology and neurometabolic investigations did not identify a diagnosis. Two liver biopsies and comparative genomic hybridisation oligo array for detailed chromosome analysis were performed and also failed to provide an answer.
Testing
The affected child and parental samples were referred for trio exome sequencing by the clinical genetics team at St George’s Hospital, London. The family history included an older brother with autism and a history of seizures in childhood that have now resolved, as well as a healthy sister. Both parents were understood to be unaffected. Single nucleotide variant (SNV) and structural variant (SV) analysis was performed using Congenica genomic data analysis software to analyze virtual panels.
Diagnosis
“Even sophisticated genetic investigations may miss a diagnosis.”
McNiven et al, 2021
Joint variant calling and annotation of SNVs and SVs was undertaken in the Congenica genomic data analysis software. No clinically relevant variants were found by SNV analysis but review of SVs showed a biparentally inherited deletion in the SCYL1 gene. Crucially, this gene is not well covered by most oligo and SNP arrays.
A recent publication [McNiven et al.] reported a multiple exon deletion in SCYL1 that was not identified by exome sequencing and the author emphasized that even sophisticated genetic investigations may miss a diagnosis. They state that a genetic diagnosis is crucial for the management of patients with acute liver failure to prevent unnecessary organ transplantations.
However, genetic testing and genomic data interpretation using Congenica resulted in the clinicians being able to identify a diagnosis of CALFAN syndrome due to a novel deletion in SCYL1. The boy was found to be homozygous for a novel deletion of 6 exons of the gene which is associated with autosomal recessive spinocerebellar ataxia-21, also known as low γ-glutamyl-transferase cholestasis, acute liver failure, and neurodegeneration (CALFAN) syndrome (MIM# 616719). The variant was shown to be biparentally inherited. 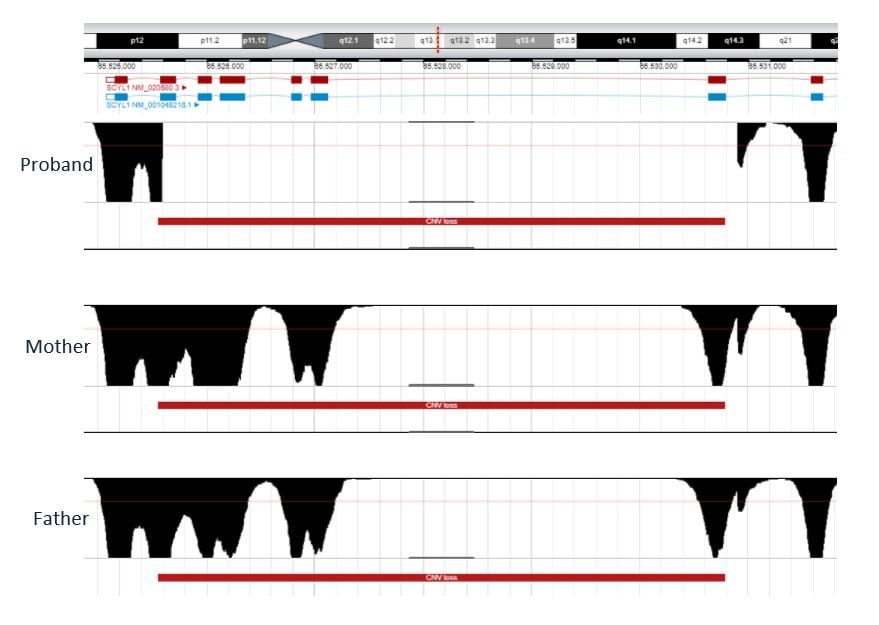 Figure 1: Coverage of the SCYL1 gene in the proband and parents. The red bar indicates that a structural variant has been detected. There is a complete absence of sequencing reads across 6 exons in the proband indicating a homozygous deletion in this region. Both parents have sequencing reads in this region but approximately half the reads that would be expected, indicating a heterozygous deletion.
Figure 1: Coverage of the SCYL1 gene in the proband and parents. The red bar indicates that a structural variant has been detected. There is a complete absence of sequencing reads across 6 exons in the proband indicating a homozygous deletion in this region. Both parents have sequencing reads in this region but approximately half the reads that would be expected, indicating a heterozygous deletion.
How is CALFAN syndrome treated?
Unfortunately, no treatment is currently available for this condition. It is a phenotype with prominent, infantile-onset hepatic involvement and variable neurological phenotype of later onset [Lenz et al.].
Importantly, in a vast majority of patients with CALFAN syndrome, acute liver insufficiency can improve without requirement of a liver transplant. Therefore, this diagnosis allows ongoing liver function surveillance and will help guide the decision making and management of this patient.
Impact
The genomic analysis using Congenica software led to a single unifying diagnosis for the child’s apparently separate medical problems after years of investigations and uncertainty. As a result, unnecessary further procedures and tests can be avoided. If exome analysis had been requested earlier the result could have avoided two inconclusive liver biopsies. The diagnosis has also been crucial for management of the child’s liver phenotype and prevention of unnecessary interventions. No further tests are now needed to understand the liver impairment.
Outcome and Future
Patients who have so far been diagnosed with this disorder have a variable phenotype, so it is hard to predict the clinical course of this condition. As the variant has been biparentally inherited, there is a 1 in 4 (25%) risk of recurrence in any future pregnancy. Therefore, this diagnosis enables informed reproductive choice and prenatal diagnosis could be considered, if appropriate.
This variant would likely have been missed by other tests as indicated by the referring clinician. Congenica software’s ability to simultaneously identify both causal SNVs and SVs ensures such cases are detected in a timely fashion and provide life-changing clinical outcomes and pathways for the patient and his family.
Try Congenica today
Start providing life-changing answers from NGS data, automatically, with the highest efficiency, accuracy and confidence.

References
McNiven V, Gattini D, Siddiqui I, Pelletier S, Brill H, Avitzur Y, Mercimek-Andrews S. SCYL1 disease and liver transplantation diagnosed by reanalysis of exome sequencing and deletion/duplication analysis of SCYL1. Am J Med Genet A. 2021 Jan 14. doi: 10.1002/ajmg.a.62079. Epub ahead of print. PMID: 33442927.
Lenz D, McClean P, Kansu A. et al. SCYL1 variants cause a syndrome with low γ-glutamyl-transferase cholestasis, acute liver failure, and neurodegeneration (CALFAN). Genet Med. 2018;20:1255–1265
.png?width=320&height=192&name=Untitled%20design%20(8).png)
.png?width=320&height=192&name=Webinar%20the%20cost%20of%20rare%20disease%20(2).png)
.png?width=320&height=192&name=Patient%20stories%20(2).png)