
Life-Changing Answers - Congenica Genomics Clinical Case Series
This clinical case study shows how genomic data analysis has expanded our understanding of rare diseases of bone development.
A paper in the European Journal of Medical Genetics (1) demonstrates the effectiveness of genomic data analysis in identifying the underlying cause of a rare and lethal skeletal disorder. It outlines how the diagnosis revealed a new clinical presentation of an established disorder, adding to our knowledge of these complex conditions and ending an eight-year diagnostic odyssey for the family.
Background on skeletal dysplasia
Skeletal dysplasias are a group of rare genetic disorders affecting bones and joints which disturb the growth and development of affected children. The disorders cause abnormally shaped bones, especially of the head and spine, and children with skeletal dysplasia often have disproportionately short arms and legs.
Skeletal dysplasias are present at birth and may even be detected in the prenatal period during routine ultrasound scans. Although individually rare, collectively they represent approximately 5% of all congenital anomalies and the birth incidence is almost 1/5000 (2). There are many different genes associated with these conditions and clinical findings are often non-specific, so accurate diagnosis can be challenging. Until the last few years treatments for these conditions have been limited and focused on surgery to manage life-threatening complications, but recent development of therapeutics has highlighted the need to be able to distinguish between the different underlying causes of skeletal dysplasia and provide accurate and timely diagnosis. (3)
Case history
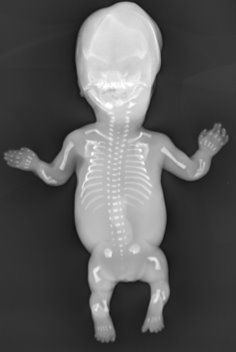
This case report describes a family with a lethal form of skeletal dysplasia diagnosed very early in pregnancy. At the mother’s routine first trimester (12 week) ultrasound examination it was noticed that the male fetus had a very small chest and short and abnormally shaped bones in the arms and legs. Based on detailed expert pathological and radiological examinations an initial diagnosis of thanatophoric dysplasia type 1 was suggested, although it was also noted that the thymus had very marked lymphocyte depletion, which is not a characteristic of this disorder.
Thanatophoric dysplasia is a lethal condition caused by DNA variants in the FGFR3 gene. It usually occurs de novo, and so the chances of a recurrence in the family was predicted to be very low.
A second affected pregnancy led to analysis of the FGFR3 gene which excluded the initial diagnosis of thanatophoric dysplasia. In total the family had three affected male pregnancies with identical clinical presentation, with the cardinal features being a very small chest, short and arms and legs, bowed thigh bones and a small thymus with marked depletion of lymphocytes.
In order to obtain an accurate diagnosis, the physician caring for the family decided to undertake exome sequencing with Congenica. Using a targeted analysis approach via applying a virtual gene panel (Genomics England PanelApp Skeletal Dysplasia panel) and variant prioritisation by Exomiser the causal variants responsible for the condition in this family were rapidly identified in the RMRP gene confirming a diagnosis of an early prenatal presentation of cartilage-hair hypoplasia / anauxetic dysplasia spectrum disorder.
Background on RMRP disorders
Cartilage-hair hypoplasia (CHH) / anauxetic dysplasia spectrum comprises of a group of autosomal recessive disorders characterised by skeletal dysplasia plus a range of additional clinical features. Both conditions are caused by variants in the RMRP gene involved in ribosomal assembly, cyclin-dependent cell cycle regulation and possibly maintenance of telomere length (4-5). Unusually this gene encodes an RNA molecule and is not translated into protein.
Cartilage-hair hypoplasia is characterised by short stature, joint laxity and a variety of haematological anomalies such as immunodeficiency, anaemia, macrocytosis, neutropenia and lymphopenia. Individuals with CHH are also at increased risk of malignancies. The signs and symptoms of CHH can vary considerably in occurrence and severity even within families.
Anauxetic dysplasia is at the more severe end of the clinical spectrum with severe short stature, dental problems, mild intellectual disability and joint laxity. The other symptoms which are typical of the overlapping condition, CHH, such as immunodeficiency and sparse hair are absent in cases of anauxetic dysplasia.
Genomic data analysis: confirming the diagnosis and expanding the spectrum
All three cases in the family were shown to be compound heterozygous for the n.64C>T and n.71 A>G pathogenic variants (also known as n.63C>T and n.70A>G). Both of these variants have been previously described in multiple families with CHH (6-9) and functional evidence indicates that they impair RMRP RNA and cyclin B2 mRNA cleavage activity (4).
The small size of the thymus and the lymphocyte depletion identified in the three cases from this family are also supportive of a diagnosis of Cartilage-hair hypoplasia rather than anauxetic dysplasia. Whilst CHH is known to have a highly variable presentation there have only been two previous reports of prenatal presentation to our knowledge (10-11), both detected by ultrasound in the second trimester of pregnancy (23 and 18 weeks respectively). Both exhibited short limbs and bowing of the thigh bone, but neither was noted to have the significant narrowing of the chest observed in this family.
It was concluded that the two known casual variants in the RMRP gene resulted in an unusually severe lethal form of cartilage-hair hypoplasia / anauxetic dysplasia spectrum disorder in the three sibling fetuses. The very early presentation, detected by ultrasound at 12 weeks’ gestation, along with major skeletal findings would place them at the severe end of the disorder spectrum, towards anauxetic dysplasia. However, given that the small thymus and lymphocyte depletion is indicative of CHH rather than anauxetic dysplasia, the fetuses were therefore be classified as severe CHH.
Impact and outcome
The diagnosis not only expands our understanding of the clinical spectrum of disorders associated with this gene, enabling other physicians to recognise and diagnose similar cases earlier, but also allows improved prognostication to inform clinical decision making.
For the family themselves, having a confirmed diagnosis after an eight-year odyssey has allowed for greater understanding of the underlying cause of the condition in their family and enabled them to make informed reproductive choices. They have since added a healthy daughter to their family.
References
- Christine M. Hall, Becky Liu, Andrea Haworth, Laura Reed, Jeremy Pryce, Sahar Mansour. Early prenatal presentation of the cartilage-hair hypoplasia / anauxetic dysplasia spectrum of disorders mimicking recurrent thanatophoric dysplasia. Eur J Med Genet. 2021 Feb 7;64(3):104162
- I M Orioli, E E Castilla, J G Barbosa-Neto. The birth prevalence rates for the skeletal dysplasias. J Med Genet. 1986 Aug;23(4):328-32
- Pauline Marzin and Valérie Cormier-Daire. New perspectives on the treatment of skeletal dysplasia. Ther Adv Endocrinol Metab. 2020. Mar 3.
- Thiel, C. T., Mortier, G., Kaitila, I., Reis, A., Rauch, A. Type and level of RMRP functional impairment predicts phenotype in the cartilage hair hypoplasia-anauxetic dysplasia spectrum. Am. J. Hum. Genet. 2007 81: 519-529,
- Svetlana Kostjukovits , Sofie Degerman , Minna Pekkinen , Paula Klemetti , Mattias Landfors , Göran Roos , Mervi Taskinen , Outi Mäkitie. Decreased telomere length in children with cartilage-hair hypoplasia. J Med Genet. 2017 May;54(5):365-370
- Bonaf ́e, L., Schmitt, K., Eich, G., Giedion, A., Superti-Furga, A.,. RMRP gene sequence analysis confirms a cartilage-hair hypoplasia variant with only skeletal manifestations and reveals a high density of single-nucleotide polymorphisms. Clin. Genet. 2002 61, 146–151.
- Ridanp ̈a ̈a, M., Jain, P., McKusick, V.A., Francomano, C.A., Kaitila, I.,. The major mutation in the RMRP gene causing CHH among the Amish is the same as that found in most Finnish cases. Am J Med Genet C Semin Med Genet 2003; 121, 81–83.
- Bonaf ́e, L., Dermitzakis, E.T., Unger, S., Greenberg, C.R., Campos-Xavier, B.A., Zankl, A., Ucla, C., Antonarakis, S.E., Superti-Furga, A., Reymond, A.,. Evolutionary comparison provides evidence for pathogenicity of RMRP mutations. PLoS Genet. 2005; 1e47, 445–454.
- Kuijpers, T.W., Ridanpa ̈a ̈, M., Peters, M., de Boer, I., Vossen, J.M., T Pals, S., Kaitila, I., Hennekam, R.C.M.,. Short-limbed dwarfism with bowing, combined immune deficiency, and late onset aplastic anaemia caused by novel mutations in the RMPR gene. J. Med. Genet. 2003, 40, 761–766.
- Crahes, M., Saugier-Veber, P., Patrier, S., Aziz, M., Pirot, N., Brasseur-Daudruy, M., Layet, V., Fr ́ebourg, T., Laquerri`ere, A., Foetal presentation of cartilage hair hypoplasia with extensive granulomatous inflammation. Eur. J. Med. Genet. 2013. 56, 365–370.
- Lam, A.C.F., Chan, D.H.C., Tong, T.M.F., Tang, M.H.Y., Lo, S.Y.F., Lo, I.F.M., Lam, S.T.S.,. Metaphyseal chondrodysplasia McKusick type in a Chinese foetus, caused by novel compound heterozygosity 64T>A and 79G>T in RMRP gene. Prenat. Diagn. 2006 26, 1018–1020.
.png?width=320&height=192&name=Untitled%20design%20(8).png)
.png?width=320&height=192&name=Webinar%20the%20cost%20of%20rare%20disease%20(2).png)
.png?width=320&height=192&name=Patient%20stories%20(2).png)