
Achieving the highest diagnostic yield possible
Genomic sequencing has the potential to rapidly analyze critically ill babies and children and also to predict a response to infectious diseases and potential therapeutic compounds. However, the identification of just one or two mutations from potentially millions of variants can take between 20 and 40 hours – even when performed by highly skilled staff using sophisticated tools.
To overcome this challenge, the variant prioritization tool Exomiser has been developed and integrated into the Congenica platform. It enables geneticists and researchers to perform faster, more accurate genomic analysis and improve diagnostic yield.

Standard exome analysis is limited
Standard exome analysis starts with a whole genome and removes off-target and common variants. The rarest variants are then prioritized based on allele frequency and pathogenicity. However, this approach rarely leads to a single, striking candidate. Exome sequencing programs have used other useful approaches too, but they are not always possible for rare disorders.
Watch Damian Smedley, Reader of Computational Genomics at the Queen Mary University of London, explain how Exomiser’s phenotype-based approach to rare disease variant prioritization is used in analysis and discovery pipelines to support better outcomes.
How does Exomiser work and why should you be interested?
Prevailing genomic pipelines leverage only a tiny fraction of the available data. Exomiser brings in under-utilized data to improve the prioritization of likely causal variants, e.g., by looking at reference knowledge of genotype to phenotype associations and how they relate to what we are seeing in the individual. In addition, Exomiser takes into account other omics-technologies such as transcriptomics and also the environmental impact of the disease and enables precise ‘fuzzy’ phenotype matching using semantic algorithms that match genetic profiles between individuals. Finally, another key aspect is leveraging the knowledge that exists on genotype-to-phenotype-associations in other species.
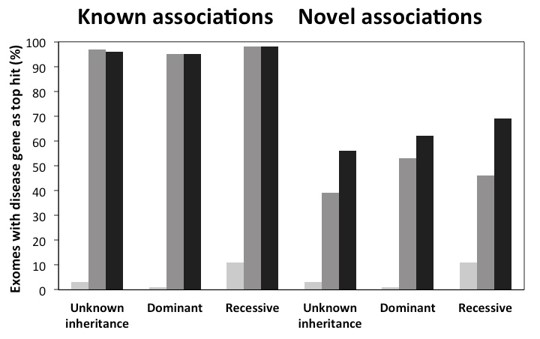
Figure 1: Benchmarking Exomiser performance on simulated exomes
Figure 1 highlights the performance of Exomiser on simulated exomes where a known disease variant has been spiked into an unaffected exome from the 1000g project. The vertical axis shows the percentage of the exomes have the causative gene as the top hit after running through Exomiser.
The first bar shows the performance we get from just using variant allele frequency and pathogenicity prioritization, the second bar shows the performance sorting by phenotypic similarity and the third bar is the combined final performance. As you may expect, for detecting known disease-gene associations, the human disease phenotype data is critical, and we can get almost 100% performance. We can also test the ability of Exomiser to discover novel associations by removing these known dis-gene associations for each run. Here the combination of variant data and phenotype is critical with the model organism and PPA data adding a lot to the performance relative to just using human disease phenotypes alone.
Increase diagnostic yield with class-leading variant prioritization
Applying Exomiser to unsolved cases enables new discoveries
Exomiser is used in the NHS undiagnosed disease program. As expected, use of genotype, phenotype and inheritance data together provided the best prioritization and was able to solve half the cases, e.g. the association between York platelet syndrome and STIM1. Regarding non-coding mutations, Exomiser uses machine learning to create a new score for whole-genome analysis. In comparison to other phenotype-based variant analysis software (PhenIX, PhenGen, eXtasy, Phevor) Exomiser performed significantly better than all other solutions [1], as shown in figure 2.
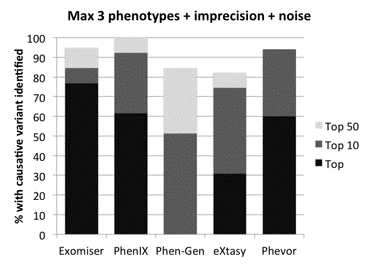
Figure 2: Comparison to other phenotype-based variant analysis software
Exomiser has been adopted by numerous research groups and laboratories and is the only approved software for variant prioritization by the International Rare Disease Research Consortium (IRDiRC).
To discover new cases for very rare conditions, Matchmaker exchange is used by researchers worldwide and Exomiser forms the phenotype matching component for some of the most successful implementations of it (PhenomeCentral and matchbox). It has also been incorporated into integrated genotype and phenotype platforms such as Phenopolis, SOLV-RD and RD-Connect. More than 20 papers have been described over the last few years describing Exomiser discoveries.
Exomiser enabled numerous independent and semi-independent confirmations in a clinical setting. For example, in a recent study that has been published on a set of 134 retinal cases that had undergone whole-genome sequencing, causal variants were identified as the top candidate in 74 % of cases and the top 5 in 94 % of cases [2].
Playing a crucial role in the 100,000 Genomes project
The 100,000 genomes project, set up by the UK government and NHS England, had the goal of sequencing of the 100,000 genomes of rare disease and cancer patients to bring the benefits of personalized medicine to the NHS, build a biomedical infrastructure and stimulate the genomics industry in the UK. The project was completed at the end of 2018 and Congenica was one of three Clinical Decision Support platforms used. Based on their success, they were selected as the sole platform for the subsequent NHS Genomic Medicine Service.
Exomiser is also part of the ISO accredited Genomics England pipeline. Exomiser is continually developed and will include further improvements regarding non-coding variants, structural variants, and new disease gene discovery to solve more cases.
Exomiser in Congenica
Exomiser has been integrated into Congenica by its original developers, Dr Damian Smedley and Dr Jules Jacobsen, for optimized variant prioritization within an end-to-end solution for secondary and tertiary genomic analysis. The software can greatly speed up the variant identification process and enables more efficient genomic analysis, with greater confidence, achieving a higher diagnostic yield than other solutions across all sample types [3,4].
Key benefits:
- Causal variants ranked 1st more than in any other tool
- Automate variant prioritization in all sample types from genomes to panels
- Complete whole genome variant prioritization in under 5-minutes
- The only software for variant prioritization approved by IRDiRC
Increase diagnostic yield with class-leading variant prioritization
Learn more about Exomiser and discover how to achieve the highest diagnostic yield possible in our Exomiser White Paper.
References
[1] Smedley D, Robinson PN. Phenotype-driven strategies for exome prioritization of human Mendelian disease genes. Genome Med. 2015;7(1):81. doi:10.1186/s13073-015-0199-2
[2] Cipriani et al., An Improved Phenotype-Driven Tool for Rare Mendelian Variant Prioritization: Benchmarking Exomiser on Real Patient Whole-Exome Data. Genes 2020, 11, 460; doi:10.3390/genes11040460
[3] Smith et al., Clinical Application of Genome and Exome Sequencing as a Diagnostic Tool for Pediatric Patients: a Scoping Review of the Literature. Genet Med. Genetics in Medicine (2019) 21: 3–16. doi: 10.1038/s41436-018-0024-6.
[4] Yang et al., Genetic aetiology of early infant deaths in a neonatal intensive care unit. Journal of Medical Genetics (2019) 0, 1 – 9
http://dx.doi.org/10.1136/jmedgenet-2019-106221


