
As part of our Patient Advocacy and Engagement program, we welcome guest blogger Ben Clay, whose daughter Tilly was diagnosed with SCN8A just before her ninth birthday.
Read their story below.

Tilly was born in Liverpool in Summer 2010. A beautiful 8lb baby girl to join her excited two-year old brother. For those first few months, despite having a new baby, everything seemed easy, almost perfect.
Exactly four months old, everything changed. Lying in her cot one morning, Tilly made a loud groan, her face froze in a grimace - jaw clamped, spit bubbling and body rigid. Neither Tilly’s mum or I had witnessed a seizure before and assumed Tilly was choking. No longer taking breaths, she was turning blue around her lips and eyes. While her desperate mum phoned for an ambulance, I dangled her upside down, banged her back, stuck my fingers down her throat to clear her airway (not advised seizure care!). Then, moving out to the street, Tilly let out an almighty gasp and breathed again. The ambulance pulled up, the first of many we would see over the years.
A&E doctors speculated it had likely been a febrile convulsion. We returned briefly to our normal life.

One month later the seizures returned with a vengeance. This time we stayed in hospital for weeks as she continued to seize multiple times each day. An MRI, CT and Lumbar Puncture all failed to diagnose a cause, but an EEG led her neurologist to suspect a metabolic disorder with an extremely poor prognosis of weeks or months to live. We left hospital devastated but the tests came back negative, the phenobarbital was helping the seizures and by the time she turned one we were feeling more optimistic.
A month after her first birthday the seizures returned and we were back in hospital. Our lives became chaotic, Tilly’s seizures regular and dangerous. New medications being tried were generally ineffective and years later we discovered one of them was known to aggravate seizures in SCN8A patients.
Reaching diagnosis
When Tilly was two, after a prolonged seizure and hospital admission, we changed her neurologist to the one on-call. He listened to our own internet research and agreed the explanation could be genetic, such as Dravet Syndrome (SCN1A) or similar. At this point SCN8A was only just being discovered across the Atlantic and Tilly was not tested for it.
Tilly tested negative for SCN1A but was given a clinical diagnosis of Dravet that allowed us to access different medications, support and a community of people going through similar experiences.
When Tilly’s neurologist retired, he wrote to his successors requesting that Tilly be tested for any new genetic epilepsies discovered, but she was not. We had been warned that finding a genetic cause was the ultimate needle in the haystack.
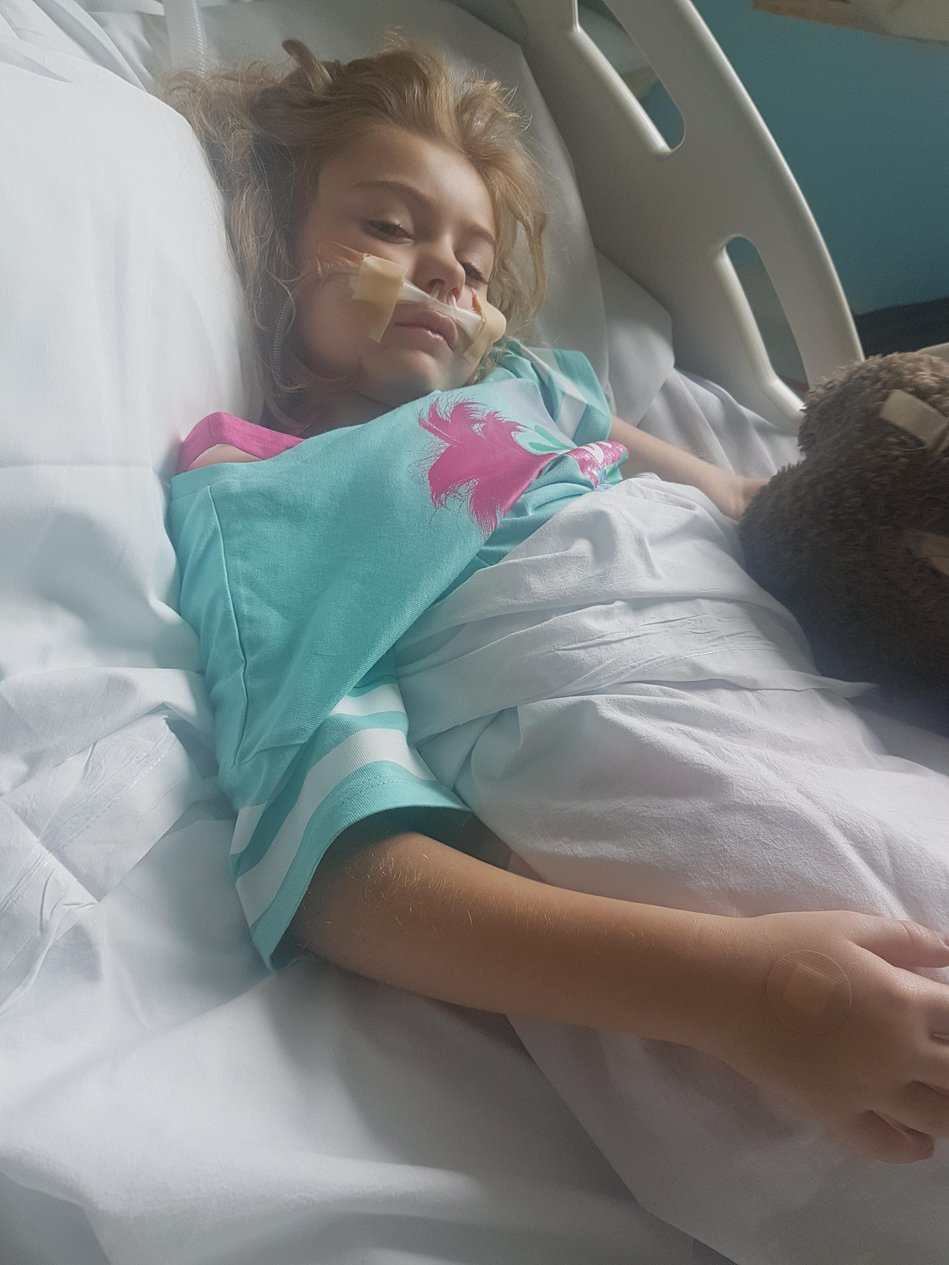
Over the years, seizures and the constant threat of seizures clouded our lives. Amplifying the anxiety was that Tilly was extremely sensitive to the ‘rescue’ medication used to stop prolonged seizures, with occasions when it suppressed her breathing. After seeing Tilly resuscitated, each seizure felt like a game of Russian Roulette and each morning our first thought was (and still is) to check Tilly was still with us.
Tilly’s development stalled, she was diagnosed with autism, and we had more hospital trips with seizures or related injuries, such as a nasty head injury or a broken arm, along with serious pneumonia.
There was a peculiar isolation of not only having a child with learning difficulties, autism and severe epilepsy but having no real explanation. We valued the connection with the Dravet Syndrome UK community but always knew it was not technically Dravet.
We speculated torturously over what it could be, if we had somehow caused it, or passed it on.
We decided not to have any more children.
Genomic testing delivers diagnosis
In 2018 a friend who worked as a genetic counsellor at Liverpool Women’s Hospital (where Tilly was born) encouraged us to participate in the 100,000 Genomes Project.
In June 2019, a month before Tilly turned nine, we received a diagnosis – a de novo variation in SCN8A. A life-changing diagnosis as children with SCN8A responded well to Sodium Channel Blockers, medications that we had always been told to avoid because of the clinical Dravet diagnosis.
We could find no information in the UK on SCN8A but did find a Facebook group – the Cute Syndrome Foundation in the USA.
In December 2019 I arrived at the Cute Syndrome conference in Baltimore. Being part of a community and hearing such similar experiences was amazing. Over 3,000 miles away, in a city I only knew from watching The Wire, I felt like I had come home.
Cute Syndrome founder, Hilary Savoie, showed that by co-ordinating the community (and working relentlessly) it was possible to have influence and attract interest from researchers and pharmaceutical companies. I also met Dr Hammer a geneticist who incredibly had identified the gene in his own daughter, Shay, soon after her tragic death in 2011, when Tilly was just a baby.
Whilst in Baltimore, I made friends with fellow Europeans including Cinzia from Italy and mum to Aron, a beautiful baby with SCN8A, and Roland from Munich who had tragically lost his gorgeous son Bruno to sudden unexpected death in epilepsy (SUDEP) aged 7. I returned to England inspired by the people I had met and those suffering or who had been lost to SCN8A.
Two months later Roland and Cinzia visited us in Liverpool and we began to plan a European network, Europe’s first SCN8A conference is due to be held in September 21, led by SCN8A Italia, University Hospital of Bonn and Danish Epilepsy Centre.
SCN8A is a sodium channel gene that plays a key role in neurology. The disorder varies in the severity of its impact, with an elevated risk of SUDEP. Research momentum is building exploring phenotype and genotype relationships and gain-of-function and loss-of-function varieties. We are also placing hope in forthcoming trials of specific SCN8A targeted medication but know more must be done.
Launching SCN8A UK & Ireland
Having the diagnosis meant we also started to find other UK families online. There are not many of us – fifteen or so that we know of, but in early 2021 our small charity – SCN8A UK & Ireland was born. Our aims are to support and connect families, so they are not alone, to provide access to information and connections to local and international expertise and to make it easier for researchers to connect to the community.
The first year after diagnosis was challenging for Tilly as we weaned existing medications and tried new ones that she was allergic to. Just before her 10th birthday she was having multiple daily seizures, she could barely walk and was drooling constantly but the day she started Lacosamide the tonic-clonic seizures stopped and she bounced back. Lacosamide would never be used for Dravet syndrome.
There is no saying how long it will last, seizure control with SCN8A is extremely difficult and many of our group continue to suffer badly, but for now it is the greatest gift we could have. Despite (or because of it all) Tilly is an amazing sparkling character with an irrepressible sense of humour, she loves singing and blowing bubbles and doing the conga. We can’t wait to meet up with the other SCN8A families.

Patient Advocacy and Engagement at Congenica
A desire to improve the lives of people living with rare and inherited diseases is central to everything we do at Congenica, and our patient advocacy and engagement initiative aims to ensure the patient voice is heard loud and clear inside the company.


.png?width=320&height=192&name=Add%20a%20title%20(2).png)
.png?width=320&height=192&name=Untitled%20design%20(8).png)