..png)

Congenica welcomes guest blogger Nuala Summerfield, founder of the Schinzel-Giedion Syndrome Foundation, who talks about her daughter Ophelia's diagnostic odyssey. The resulting genomic diagnosis has had a life-changing impact on their lives.
Early signs
My daughter Ophelia was born in 2011 in UK after an uneventful pregnancy. Labour was induced at 40 weeks as an ultrasound scan showed a sudden decrease in the amount of amniotic fluid
surrounding her. I gave birth in a London teaching hospital, which in hindsight was incredibly
fortunate, as although the labour was uneventful, Ophelia suffered a respiratory arrest within
minutes of birth. Thankfully there were a team of neonatal specialists on hand to resuscitate her, but she was critically ill and spent the first three weeks of her life in the Neonatal Intensive Care Unit (NICU).
.jpg?width=584&name=Ophelia%20newborn%20(2).jpg)
Both in the NICU and during the subsequent months after Ophelia was able to come home from hospital, it was very clear that she was not developing typically. Ophelia had problems feeding as she had a weak suckle, difficulty swallowing and frequent reflux. She was very irritable and cried a lot, she did not like to be held and seemed to find it difficult to sleep. She also did not appear to respond to visual and sound stimuli, so we were unsure if she could see or hear.
The multiple medical specialists that we were referred to strongly suspected that Ophelia had a genetic condition but were uncertain which one. Several genetic syndrome names were suggested but as the symptoms that Ophelia showed were quite non-specific, no genetic testing was performed.
Genetic testing leads to diagnosis
When Ophelia was nine months old, she developed infantile spasms, a serious form of epilepsy. At the time we were living in Switzerland and the paediatric neurologist there referred us to a clinical geneticist who ordered a genetic testing panel for the 50 genes most frequently associated with infantile epilepsy. By some stroke of luck or possibly fate, this geneticist had actually trained under Professor Schinzel (of Schinzel-Giedion Syndrome) and although he had never met a child with this ultra-rare genetic condition, he had a strong suspicion based on what he knew about the syndrome and Ophelia‘s appearance and symptoms that this could be her genetic diagnosis.
His hunch was correct, and we received confirmation that Ophelia had a de novo mutation affecting the SETBP1 gene, which causes Schinzel-Giedion Syndrome (SGS). SGS is a very severe, life-limiting condition affecting many body systems. The SETBP1 gene, which is located on chromosome 18, provides instructions to cells on how to make SETBP1 protein, which we are now discovering is extremely important for normal brain development as well as in the development of many other organs.
At the time that we received the diagnosis in early 2012 there was no patient group for SGS and no patient-friendly information available about SGS on the internet. There was no active research
focusing on SGS, just a handful of technical medical genetics publications. When we were informed of Ophelia’s diagnosis, we were told that based on the available knowledge from these few medical publications, her life expectancy was three - four years and that we should go home and enjoy the time that we had with her.
Lots of questions and no answers
Like all parents of children with rare and ultra-rare disease, getting genetic conformation of a diagnosis was a very important step for us. However, it still left us with so much uncertainty and many unanswered questions in terms of what this diagnosis meant for Ophelia and for us as her family. How could we ensure that she received the optimal medical care and therapies when so little was known about the condition?
This is a common problem in rare and ultra-rare disease – how do doctors develop expertise and knowledge of these conditions when that child may be the only patient they will ever treat with that specific condition?
Fewer than 100 children worldwide have been genetically diagnosed with SGS, but the true
incidence is unknown. There are five children living with SGS in the UK that we know of currently.
SGS is a multisystemic condition as the specific SETBP1 mutations affect the development of many body systems including the brain, gastrointestinal, urinary and respiratory tract. SETBP1 is also an oncogene and children with SGS are also at increased risk of developing certain types of cancer.
Ophelia's symptoms and treatment
Like almost all children with SGS, Ophelia has severe medically refractory epilepsy and severe global developmental delay. Since she was first diagnosed with epilepsy at nine months old, she has been treated with most of the available seizure medications and the ketogenic diet, but her seizures have persisted despite medical management. Her seizure type has varied frequently over the years and currently she has daily tonic and tonic-clonic seizures.
Ophelia has difficulty sleeping and has severe developmental delay and learning difficulties. We have discovered that she has cortical visual and hearing impairment which explains the difficulty that she has interacting with her environment, but with the correct professional help she is now making good improvement. She is non-verbal, but communicates with us through sounds and body language and she certainly knows what she likes and what she doesn’t!
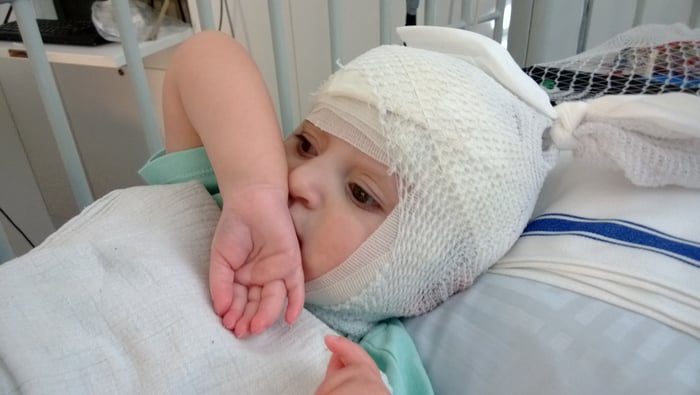
Pre-Covid, Ophelia attended a special needs school daily where she had a 1:1 highly trained support teacher and received daily physiotherapy, occupational therapy and speech and language therapy together with multi-sensory impairment support and hydrotherapy. These therapies are so important to help her to interact with her environment and to maintain the comfort and range of movement of her joints since she is not mobile. As a family, we have been hugely impacted by the pandemic, as Ophelia is deemed extremely clinically vulnerable and so has been shielding at home with us since March 2020, unable to attend school or access her therapies. Thankfully Ophelia has just started back at her school, but the impact of missing a whole year is considerable.
The importance of early diagnoses
SGS impacts many organ systems and therefore has a profound effect on both the affected child and their families who need to provide 24/7 care for them. However, it has become clear to me from my experience of caring for Ophelia, that an early diagnosis and the appropriate intervention and support from relevant medical and healthcare professionals can have a profound effect on quality of life and outcome for children born with this rare disease. Ophelia‘s life expectancy according to the previously published sparce medical literature was less than four years old.

Ophelia has recently celebrated her 10th birthday, so although many children with SGS sadly do not do as well as she has, it proves that there is a spectrum of disease that is wider than first appreciated.
Children born with SGS have a very characteristic facial phenotype, which if recognised could prompt genetic testing in the postnatal period and help accelerate the time to diagnosis, thus shortening the diagnostic odyssey and improving outcomes for both SGS children and their families.
Establishing the Schinzel-Giedion Syndrome Foundation
As recently as two years ago there were no active programmes of scientific research focused on SGS. For families of children with SGS and the medical professionals looking after these children, this was a desperate situation. This desperation and lack of hope was very evident from the many discussions and comments on our private Facebook Group from SGS families all over the world.
As a rare disease community, we felt that we were ‘too rare’ to be of interest to scientists and Pharma, and without research interest there is no opportunity to advance knowledge of a rare condition and no hope for the development of new, effective targeted treatments for our children.
This was the motivating force for establishing The Schinzel-Giedion Syndrome Foundation as a
registered UK charity in November 2019. Our charity is run by an international group of volunteer
Trustees and all of us are parents of children with SGS. The SGS foundation was created to represent children with SGS and their families living around the world and this is the only charity representing this rare genetic condition. Our mission is to provide support to affected families, to raise awareness of SGS particularly within the medical profession, and to facilitate and support scientific research.
Our vision is for a future in which all children born with SGS will receive a rapid genetic diagnosis and have access to effective targeted medical and gene therapies to ensure that they live longer,
healthier and happier lives.
Delivering results on a personal and professional level
As a charity we are very fortunate to be supported by an expert scientific and medical advisory
board (SMAB). We are working with our SMAB to launch our international SGS patient registry, to
develop Standard of Care Guidelines for SGS and to develop our research strategy.
We have focused on engaging with leading international researchers to generate interest in SGS and the causative SETBP1 mutations as an area of research interest in both the fields of epilepsy / neuro developmental disorders but also oncology. All this hard work has paid off and has resulted in over €2M of European funding being awarded to researchers to focus on the development of new epilepsy treatments for our children. This is the first major research funding awarded specifically for SGS and the hope is that this research will make discoveries that will also benefit other rare epilepsies which may share similar seizure mechanisms to SGS.
As SGS is such a multi systemic disorder, we now need to focus on developing similar collaborations with interested research groups in the fields of urology /nephrology and oncology, so we can work together to secure research funding and keep advancing the science of SGS / SETBP1 forwards. I am very proud of the amazing progress that the Foundation has made for SGS over the past 18 months, which is testament to the commitment and effort from our Trustees, our SMAB, our research collaborators and our families.
.jpg?width=486&name=Ophelia%202016%20(2).jpg)
Having a child with an ultra-rare genetic condition is certainly not without its challenges but I feel
incredibly fortunate to have Ophelia in my life. She has taught me so much about patience, unconditional love and appreciation of the small but important things. Despite all of the major challenges she faces, she loves life with a passion and her amazing resilience and will-to-live is a daily inspiration to all who know and love her.
Support for rare disease patients and their families
Congenica's Patient Advocacy and Engagement team work with people across the world, helping to navigate often confusing and disparate information by providing educational materials that are trustworthy and helpful. Our aim is to ensure patients, clinicians and researchers understand the patient journey from referral, through diagnosis and beyond.
To see Nuala talking about SGS and the impact genomics has had on her family, and other patient groups talking about their experiences of rare disease, visit our Rare Disease Day 2021 blog.

To find out more about Schinzel-Giedion Syndrome, visit www.sgsfoundation.org
.png?width=320&height=192&name=Untitled%20design%20(8).png)
.png?width=320&height=192&name=Since%202016%2c%20the%20number%20of%20women%20working%20in%20STEM%20fields%20in%20the%20UK%20has%20increased%20by%20216%2c552%2c%20taking%20the%20total%20number%20over%20the%201%20million%20mark%20for%20the%20first%20time.%20Women%20now%20make%20up%2024%25%20of%20the%20STEM%20workforce%20i%20(2).png)
-1.png?width=320&height=192&name=Deciphering%20Developmental%20Disorders%20(1)-1.png)